The subroutines
The NBO transformation was implemented originally with the CNDO code in the BONDO program, and has been implemented in our group with the EH code.
HP-UX version and
Linux version for the modified BONDO program
are provided. The HP-UX version makes use of the BLAS library that
comes with the HP-UX workstation. In the Linux version, the Strassen
method for matrix multiplication is used.
The Here is a list of the (modified)
BONDO subroutines and their functionalities (the original BONDO routines
the upper case ones, our work the lower case ones):
Having read an input PDB structure, the program detects whether
or not there exist benzene molecules (BENZ), tryptophan, tyrosine,
and phenylalanine
in the system. If yes, flags In order to construct the delocalized aromatic pi bonds,
all the hybrid orbitals have to be known. Before excuting the snippet
that does the job for aromatic rings, the program has already searched
and established all the lone pairs and two-center bonds. Therefore,
up to now, every carbon atom of an aromatic ring has three known sp2
atomic hybrids that form the bonds with two nearby carbon atoms and
the dangling hydrogen atom. By use of the orthonormality conditions, the
fourth atomic orbital can be reconstructed from these three known
hybrids. Subroutine A plot that shows success of the above scheme can be found
here. The average MO energies corresponding
to the aromatic ring exhibit convincing degeneracy, proving that
the aromatic ring is still chemically an aromatic ring in the protein
environment. (Another sign that the CHARMM force field is capable of
maintaining the chemical stability of proteins.)
The cause of such (false) fluctuations became clear later on: They
are caused by the population degeneracy of the two orbitals in the doube
bonds and lone pairs. The two eigenstates obtained by diagonalizing
the density matrix subblocks corresponding to these degenerate bonds
are, in actuality, sort of random mixture of the two bonding orbitals.
Fluctuations therefore change the couplings of
these false bonds with others stochastically, with a magnitude
comparable up to twice of the absolute values of the couplings.
The effect of these false fluctuations could have been eliminated
had we taken the K matrix into integration. The K matrix will counteract
the role of these false fluctuations when the Schroedinge equation is
integrated. But as we have dropped the K matrix off in the CNDO
calculations, the false fluctuations would show off their influence
on the electronic dynamics, particularly when they are adjacent to the
donor and acceptor. The consequence is always a gradual and steady rising
of electronic population in the bridge.
Unlike the case of the aromatic rings, a C=O double bond or O lone pair
has two unknown hybrids that are needed to be determined. Obviously,
the condition of orthonormality cannot be used to construct two unknown
hybrids from the other two known ones. Therefore, we have to turn to other
methods to dedegenerate them.
One of the methods, as Spiros suggested, is to make use of the
energetical nondegeneracy of the bonds. Namely, we transfer the
determination of the bonds to the Fock space, instead of diagonalizing
the density matrix subblock, we diagonalize the Fock matrix subblock.
Because there is an energy gap between the two states in a double bond,
they can therefore be distinguished. Code:
C=O double bonds and
O lone pairs.
But does such directional flipping happen in our simulations?
Yes. This problem will be frequently encountered in our simulations.
When we solve the Roothaan equation HC=SCE, there are in principle
two solutions for the eigenvectors C or -C. There is no
criterion which one is more correct. As a convention, the component of
the eigenvector at the first position C(1) is usually assumed to be
always positive (different diagonalization subroutine may use different
convention, but it should be similiar). In a static calculation, this
is not a problem, because we never need to calculate the time derivative
of the wave functions. In a dynamic calculation, thermal fluctuations can
easily make C(1) change its sign from time to time if its magnitude
is not greater than that of the fluctuations. If all the natural bonds
use this convention to define their directions, we will see some of the
bonds that have small C(1) flipping from time to time.
The solution to such a problem can be described in the following
steps: (1) Select the first Hamiltonian in the time series, build the
natural bonds, and remember the directions of all bonds (store them
in a file In CNDO calcullations, we don't calculate the K matrix,
since we neglect the advanced and retarded overlaps as we
have neglected the punctual overlaps. Atomic adiabatic couplings
are calculated within the framework of EH. (See the section
Extended Huckel Calculation for a thorough
description about the calculation method.
© 2000, Qian Xie
input_lcbo sets
the flags for CNDO calculations on the NBO basis (also called
the Linear Combination of Bond Orbitals method).
aromatic(DM,T,N,NATOMS,ipr): Constructs the AO-BO orthogonal
transformation, the T matrix using the input density matrix
DM. This subroutine is able to handle
aromatic rings and benzene molecules. This is the main routine.
LOAD(DM,IAT1,IAT2,IAT3,BLK,NB): Loads the subblock of the
density matrix that corresponds to the three different atoms with indices
IAT1, IAT2, IAT3.
JACVEC(NB,BLK,EVAL,C,12): Diagonalization routine that
comes with the original BONDO. EVAL stores the eigenvalues.
EXTRCT(C,EVAL,BORB,OCC,NB,IRNK): Extracts eigenvector
of IRNK and stores it in BORB.
DEPLET(DM,BORB,OCC,NB,IAT1,IAT2,IAT3): Deplete the lonepairs
(or in search for the three-center bonds, the bicentral bonds) from the
density matrix.
STASH(BORB,OCC,IBD,IAT1,IAT2,IAT3): Normalizes
BORB, puts it into the correct position in the
Q matrix, and extracts the polarization coefficient.
fourth(ibd,iring,natoms,ipr): Builds the fourth atomic
orbitals of carbon atoms at aromatic rings according to the conditions of
orthonormality.
REFORM(nring,T,NBSZR,N,NATOMS,ipr): Sets up the final
form of the transformation T for all bonds except the aromatic
pi bonds.
REFORMaro(nring,T,NBSZR,N,NATOMS,ipr):
Fill the aromatic pi bonds part in the T matrix. This
is the final step for building the AO-BO transformation.
ANLYZE(T,NDIM): Analyze the T matrix,
report the polarization factors,
the p characters, and the bond orientation angles (see an example
output file bond.index).
cobond(fk,fkao,dm,t,n,ipr): Dedegenerates C=O double
bonds.
lonepair(fm,dm,n,ipr): Dedegenerates O lonepairs.
findsign(n,ipr): Remembers bond orientations of the
(reference) first conformation.
compsign(n,ipr) and
compsignaro(n,ipr): Rectifies bond orientations of the
successive conformations.
trans(n,1): Transform the Fock matrix from AO to BO
(The Fock matrix in the NBO basis is what we want after all these efforts).
How to handle aromatic rings
The original subroutine for constructing orthogonal matrix
transformation from atomic orbitals (AO) to bond orbitials (BO),
NATHYB in BONDO, was replaced by an aromatic-ring-capable
subroutine, aromatic.
aring true, and deposits the
index of the six carbon atoms into the array idc6(i,nring).
The maximum number of aromatic rings allowed is 20, as declared in the
common block for idc6. The following snippet describes
how this is done.
aring=.false.
nring=0
do iat = 1 , natoms
if(resname(iat).eq.'BENZ'.or.
: resname(iat).eq.'TYR '.or.
: resname(iat).eq.'PHE ') then
aring=.true.
if(symbol(iat).eq.' CG ') then
nring=nring+1
idc6(1,nring)=iat
endif
if(symbol(iat).eq.' CD1') then
idc6(2,nring)=iat
endif
if(symbol(iat).eq.' CD2') then
idc6(3,nring)=iat
endif
if(symbol(iat).eq.' CE1') then
idc6(4,nring)=iat
endif
if(symbol(iat).eq.' CE2') then
idc6(5,nring)=iat
endif
if(symbol(iat).eq.' CZ ') then
idc6(6,nring)=iat
endif
endif
if(resname(iat).eq.'TRP ') then
aring=.true.
if(symbol(iat).eq.' CD2') then
nring=nring+1
idc6(1,nring)=iat
endif
if(symbol(iat).eq.' CE2') then
idc6(2,nring)=iat
endif
if(symbol(iat).eq.' CE3') then
idc6(3,nring)=iat
endif
if(symbol(iat).eq.' CZ2') then
idc6(4,nring)=iat
endif
if(symbol(iat).eq.' CZ3') then
idc6(5,nring)=iat
endif
if(symbol(iat).eq.' CH2') then
idc6(6,nring)=iat
endif
endif
enddo
fourth is to do this job. If the print
level ipr is greater than 3, it will also check if the
reconstructed orbital is strictly orthogonal to the three parent orbitals.
The reconstructed orbitals are deposited in the array Q,
together with the hybrids and lonepairs. The subroutine
REFORMaro is used to construct the aromatic part of the
T matrix:
SUBROUTINE REFORMaro(nring,T,NDIM,N,NATOMS,ipr)
.
.
.
dimension anorm(6),cop(6,6)
data cop/1.0, 1.0, 1.0, 1.0, 1.0, 1.0,
: 2.0, 1.0, 1.0,-1.0,-1.0,-2.0,
: 0.0,-1.0, 1.0,-1.0, 1.0, 0.0,
: 0.0, 1.0,-1.0,-1.0, 1.0, 0.0,
: 2.0,-1.0,-1.0,-1.0,-1.0, 2.0,
: 1.0,-1.0,-1.0, 1.0, 1.0,-1.0/
anorm(1)=1.0d0/dsqrt(6.d0)
anorm(2)=0.5d0/dsqrt(3.d0)
anorm(3)=0.5d0
anorm(4)=anorm(3)
anorm(5)=anorm(2)
anorm(6)=anorm(1)
C REORDER OCCUPIED BO'S TO PUT LONE PAIRS LAST and pi bonds second last
DO 10 NLP=1,NOCCA
IF(LABEL(NLP,1).NE.LLP) GO TO 20
10 CONTINUE
20 NLP=NLP-1
NPB=0
DO NPI=1,NOCCA
IF(LABEL(NPI,1).EQ.LPB) NPB=NPB+1
ENDDO
NBD=NOCCA-NLP-NPB
do ibd = 1 , n
if(ibd.le.nlp) then
ibx(ibd)=ibd+nbd+npb
elseif(ibd.gt.nlp.and.ibd.le.nocca) then
ibx(ibd)=ibd-nlp
else
ibx(ibd)=ibd
endif
enddo
lumo=nlp+nbd+npb+1
c>>> build the part of the transformation matrix correponding to the PI bonds
do iring = 1 , nring
jet=0
do ibd = nlp+nbd+3*iring-2, nlp+nbd+3*iring
jet=jet+1
do iat = 1 , 6
jl=ll(idc6(iat,iring))
ju=ul(idc6(iat,iring))
irow=0
icol=jl+ino(idc6(iat,iring))-1
do j=jl,ju
irow=irow+1
t(j,ibx(ibd))=q(irow,icol)*cop(iat,jet)*anorm(jet)
enddo
enddo
enddo
do ibd = nocca+nbd+3*iring-2, nocca+nbd+3*iring
jet=jet+1
do iat = 1 , 6
jl=ll(idc6(iat,iring))
ju=ul(idc6(iat,iring))
irow=0
icol=jl+ino(idc6(iat,iring))-1
do j=jl,ju
irow=irow+1
t(j,ibx(ibd))=q(irow,icol)*cop(iat,jet)*anorm(jet)
enddo
enddo
enddo
enddo
.
.
.
In the above code snippet, the magenta lines define the prefactors
of linear combinations for the pi bonds and antibonds,
cop. The blue lines build the pi bonds,
namely, the linear combinations of
the renormalized atomic hybrids obtained by fourth.
Dedegenerating double bonds and lone pairs
We found extraordinarily large fluctuations of direct couplings
of C=O double bonds and O lone pairs with others
(a frequently encountered case
in amino acids calculations) when analyzing the time series of the
Hamiltonian matrix elements (This has, mistakenly, led to what Spiros
calls the electronic pumping phenonmenon, which serves as an example
that ultrafast and ultra-intensive electronic state fluctuations
could result in unusual transport-like behavior).
Avoid random orbital orientation flipping
A natural bond in the NBO method is represented by a linear combination
of the basis of a Hilbert space spanned by the atomic wave functions
s, px, py, pz of all the atoms
involved in the formation of the bond. Although a single chemical bond,
for example, a bicentral bond, has no significant importance for
its interal direction (the direction in which the bond points
from one atom to the other),
there is a problem of directionality if we have to deal with the
time dependence.
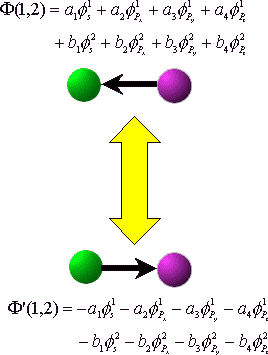
The above two bonds are equivalent, but
their orientations are different.
In our time dependence simulations on the basis of NBO,
the wave function basis at each time is reconstructed by diagonalizing
the sub density matrices. The basis, therefore, fluctuates with time.
Such fluctuations result in an electronic friction matrix which enters
the time-dependent Schroedinger equation as an imaginary peer together
with the Hamiltonian. The
magnitude of the friction matrix, numerically, is determined by how
rapidly the basis changes. If the basis varies slowly, then the friction
matrix (sometimes we call the K matrix) can be calculated more accurately.
Due to this reason, we do not hope to see the basis has drastical changes
like arbitary
directional flipping from one state to the other (shown in the above picture).
If, for instance, the bonding directions flip at the rate of one flip
per femtosecond, the situation would amount to having heavy contributions
at the high end of the Fourier spectrum, which disfavors the cubic spline
interpolation method we employ to obtain intermediate Hamiltonians between
flips (cubic splines perform well for smoothly
changing functions but very poor for oscillatory functions).
tsign or
tsign.hasAromaticRings), see subroutine
findsign(n,ipr) in
the source file fixsign.f; (2) Build the natural
bonds for successive Hamiltonians, rectify the bond directions according
to the remembered directions (stored in the file tsign),
see subroutines
compsign(n,ipr) and
compsignaro(n,ipr) in
the source file fixsign.f.
Below is an example of the direction-memory file:
1 BD 1 C 2 H 5 1 .6949
2 BD 1 C 3 H 6 1 .6924
3 BD 1 C 4 H 7 1 .6995
.
.
.
239 PB 1 135 C 326 1 .3756 136 C 330 1 .3621 138 C 335 1 .3808 140 C 340 1 .3604 142 C 345 1 .3748 144 C 350 1 .3656
240 PB 2 135 C 326 1 .5311 136 C 330 1 .2561 138 C 335 1 .2692 140 C 340 -1 .2549 142 C 345 -1 .2650 144 C 350 -1 .5171
241 PB 3 135 C 0 1 .0000 136 C 330 -1 .4435 138 C 335 1 .4663 140 C 340 -1 .4415 142 C 345 1 .4590 144 C 0 1 .0000
242 PB 1 173 C 424 -1 .3797 174 C 428 -1 .3904 176 C 433 -1 .3904 178 C 438 -1 .4000 180 C 443 -1 .3941 182 C 448 -1 .3972
243 PB 2 173 C 424 -1 .5370 174 C 428 -1 .2760 176 C 433 -1 .2760 178 C 438 1 .2828 180 C 443 1 .2786 182 C 448 1 .5617
244 PB 3 173 C 0 1 .0000 174 C 428 1 .4781 176 C 433 -1 .4781 178 C 438 1 .4899 180 C 443 -1 .4826 182 C 0 1 .0000
245 PB 1 193 C 478 1 .2999 194 C 482 1 .3006 196 C 487 1 .3079 198 C 492 1 .3134 200 C 497 1 .3193 202 C 502 1 .3282
246 PB 2 193 C 478 1 .4241 194 C 482 1 .2126 196 C 487 1 .2177 198 C 492 -1 .2216 200 C 497 -1 .2258 202 C 502 -1 .4641
247 PB 3 193 C 0 1 .0000 194 C 482 -1 .3682 196 C 487 1 .3771 198 C 492 -1 .3838 200 C 497 1 .3911 202 C 0 1 .0000
248 LP 6 O 14 1 .8780
249 LP 6 O 12 1 .8738
250 LP 7 N 17 1 .8840
.
.
.
306 BD* 1 C 2 H 5 -1 .7191
307 BD* 1 C 3 H 6 -1 .7215
308 BD* 1 C 4 H 7 -1 .7146
.
.
.
544 PB* 1 135 C 0 1 .0000 136 C 330 1 .4435 138 C 335 -1 .4663 140 C 340 -1 .4415 142 C 345 1 .4590 144 C 0 1 .0000
545 PB* 2 135 C 326 1 .5311 136 C 330 -1 .2561 138 C 335 -1 .2692 140 C 340 -1 .2549 142 C 345 -1 .2650 144 C 350 1 .5171
546 PB* 3 135 C 326 1 .3756 136 C 330 -1 .3621 138 C 335 -1 .3808 140 C 340 1 .3604 142 C 345 1 .3748 144 C 350 -1 .3656
547 PB* 1 173 C 0 1 .0000 174 C 428 -1 .4781 176 C 433 1 .4781 178 C 438 1 .4899 180 C 443 -1 .4826 182 C 0 1 .0000
548 PB* 2 173 C 424 -1 .5370 174 C 428 1 .2760 176 C 433 1 .2760 178 C 438 1 .2828 180 C 443 1 .2786 182 C 448 -1 .5617
549 PB* 3 173 C 424 -1 .3797 174 C 428 1 .3904 176 C 433 1 .3904 178 C 438 -1 .4000 180 C 443 -1 .3941 182 C 448 1 .3972
550 PB* 1 193 C 0 1 .0000 194 C 482 1 .3682 196 C 487 -1 .3771 198 C 492 -1 .3838 200 C 497 1 .3911 202 C 0 1 .0000
551 PB* 2 193 C 478 1 .4241 194 C 482 -1 .2126 196 C 487 -1 .2177 198 C 492 -1 .2216 200 C 497 -1 .2258 202 C 502 1 .4641
552 PB* 3 193 C 478 1 .2999 194 C 482 -1 .3006 196 C 487 -1 .3079 198 C 492 1 .3134 200 C 497 1 .3193 202 C 502 -1 .3282
The first and second columns are the bond indices and bond codes, listing
bonds (BD), lonepairs (LP) and pi bonds and their antibonds (except
lonepairs). For BD(*)s and LP(*)s,
from the third to the sixth, the indices and names of atoms
that participate the formation of the bonds are listed. The seventh lists
the positions and signs of the maximum componenents (absolute value,
of course). The last lists the absolute values of the maximum components.
For PB(*)s, the regulation is slightly distinct. All the six atoms,
their indices, names, positions, signs and values of the maximum components,
are listed as above.
Compute the adiabatic couplings