Prepare a structure
Brookhaven Protein Data Bank
presents a large number of cystallographic structures for proteins.
If the resolution is not high enough to reveal the hydrogen atoms
(as is the usual case),
the PDB file will not contain the coordinates for the hydrogens.
The HBUILD command has to be used to set the hydrogen
positions.
If the
size of the protein is too big for a workstation case, consideration
should be taken to cut the most important part of the protein out,
usually containing the reaction site and the interesting region.
Harmonic restraints can be used to clinch the residues which cross
the cutoff boundary. Energy minization is normally performed to
remove bad contacts in the cystallographic structure. It is
advised that the protein be equilibrated for some time with harmonic
restraints around the crystallographic positions.
Care has to be taken to establish the disulfide bonds, decide
the protonation of the residues, install the terminals,
and manage charge neutrality for
the neutral amino acids and ligands.
Force fields in molecular simulations play the decisive role on
the results. To some extent,
determination of the force fields parameters is to
quantify the potential terms in the Hamiltonian. Therefore the
quantitative results produced by a given Hamiltonian
will rest on the input of the force fields parameters, provided
that the simulation protocols are identical.
There are two important Charmm files: The structure builder
(
If the protein consists of a ligand which is not one of
the 20 amino acids, or, none of the nuts and bolts the CHARMM parameter
and topology files present, we will have to build it into
the parameter and topology files.
Definition of a component in the topology input is
composed of defining the following information:
(1) Type of atoms; (2) Charge of atoms; (3) Bonds; (4) Dihedral
angles (proper and improper);
(5) Internal coordinates; (6) Donor and acceptor of hydrogen bonds;
(7) Patches to modify the structure locally.
The following is an example for building a triple hybrid that
is composed of
three different groups ( In the lines that start with In the above example, the internal coordinates are not presented.
The coordinates are input from an PDB file which contains the Cartesian
coordinates of the hybrid. Part of the structure is for free energy
calculation (lambda dynamics), which may not be relevant for electron
transfer simulation. But yet it shows how to create a structure in
Charmm.
Once a structure is built, Charmm will ask for the corresponding
force fields, namely, the bonds, angles, and dihedrals. These parameters
have to be defined in the parameter file. For example, if one wants
to add a bond to the parameter file, just add a line like this in the
BOND category
Charmm's parameter file has parameters for biologically
important elements on many different occasions. There are elements that
may not have well tested parameter sets for the time being.
For instance, for
a complex containing metallic groups, the user usually needs to
develop his own parameter sets. FYI, a periodic table shows
how many elements we have to live with
While it is being done, an MD simulation carries rich information about
the atomic coordinates and the velocities. Considering the number
of atoms and the structure complexity involved in protein simulations,
data management and analysis become a very time-consuming job, and
sometimes, could be intimidating and frustrating for a novice.
Although it is desirable that data analysis be performed simultaneously
while the MD simulation is going on, for simulation of complicated systems
it can be very difficult to decide what we want to know at the stage of
preparing script inputs. Particularly when we are using a generic simulation
client such as Charmm that claims to be able to run any kind of MD job,
it can be sometimes awkward to manipulate the MD machine on a
developer-provided scripting environment.
A better way of working with Charmm is to deposit a long trajectory
into a file so that we can get back to this file and postprocess it at
any time. This file is normally called dynamical coordinate data (DCD),
any file with an extention name 'dcd' is recognized as an DCD trajectory
file, and can be played by the DCD player built in the VMD.
In this way, Charmm looks somehow like a DCD recorder---it records the
important information for the evolution of the system(of course,
guided by the equation of motion and the selected simulation protocols).
Once the DCD file is saved, nothing is actually lost after the simulation
is completed.
Postprocessing a simulation in Charmm
We have also written up a DCD parser
which is independent of the Charmm platform. The program takes
a DCD and two PDB files (a starting and a reference structure)
as selected in the input file and
can do some simple calculations much faster because the overheads of the
whole Charmm platform are avoided. The DCD parser can handle the following
tasks:
Define force fields
top_all22_prot_na.inp)
that contains the nuts and bolts for building proteins and nucleic acids,
and the parameter file (
par_all22_prot_na.inp) that contains the force fields
paramters. Besides loading existing structure data,
Charmm's structure builder enables the user to construct
proteins and nucleic acids from scratch.
hydan, nhydan, mhydan).
RESI HYBR 0.000 ! hybrid: hydan+nhydan+mhydan
!
GROU !
ATOM C1 CTS 0.220 ! O6-HO6
ATOM C7 CF 0.510 ! |
ATOM O7 OF -0.510 ! H61-C6-H62
ATOM N1 NH1F -0.430 ! | O7 *
ATOM HN1 HF 0.310 ! | || /
ATOM C8 CF 0.630 ! H5-C5---O5 || /
ATOM O8 OF -0.510 ! H4 / \ C7--N1
ATOM N2 NH1F -0.470 ! \ / HO2 \ / \
ATOM HN2 HF 0.310 ! C4 | C1 C8=O8
ATOM C5 CTS 0.250 ! / \ H3 O2 / \ /
ATOM H5 HAS 0.090 ! HO4-O4 \| | / N2
ATOM O5 OES -0.400 ! C3---C2 |
ATOM C7B CF 0.510 ! | | HN2
ATOM O7B OF -0.510 ! HO3-O3 H2
ATOM N1B NH1F -0.111 !
ATOM N3B NH2 -0.629 !
ATOM HN31 H 0.310 !
ATOM HN32 H 0.310 !
ATOM C8B CF 0.630 !
ATOM O8B OF -0.510 !
ATOM N2B NH1F -0.470 !
ATOM HN2B HF 0.310 !
ATOM C7C CF 0.510 !
ATOM O7C OF -0.510 !
ATOM N1C NH1F -0.220 !
ATOM C9C CT3 -0.170 !
ATOM H91 HA 0.090 !
ATOM H92 HA 0.090 !
ATOM H93 HA 0.090 !
ATOM C8C CF 0.630 !
ATOM O8C OF -0.510 !
ATOM N2C NH1F -0.470 !
ATOM HN2C HF 0.310 !
GROU !
ATOM C2 CTS 0.140 !
ATOM H2 HAS 0.090 !
ATOM O2 OHS -0.660 !
ATOM HO2 HOS 0.430 !
GROU !
ATOM C3 CTS 0.140 !
ATOM H3 HAS 0.090 !
ATOM O3 OHS -0.660 !
ATOM HO3 HOS 0.430 !
GROU
ATOM C4 CTS 0.140
ATOM H4 HAS 0.090
ATOM O4 OHS -0.660
ATOM HO4 HOS 0.430
GROU
ATOM C6 CTS 0.050
ATOM H61 HAS 0.090
ATOM H62 HAS 0.090
ATOM O6 OHS -0.660
ATOM HO6 HOS 0.430
BOND C1 O5 C1 C2
BOND C2 H2 C2 O2 O2 HO2 C2 C3 C3 H3
BOND C3 O3 O3 HO3 C3 C4 C4 H4 C4 O4
BOND O4 HO4 C4 C5 C5 H5 C5 C6 C6 H61
BOND C6 H62 C6 O6 O6 HO6 C5 O5
BOND C1 C7 C7 N1 N1 HN1
BOND N1 C8 C8 N2
BOND N2 C1 N2 HN2
BOND C7 O7 C8 O8
BOND C1 C7B C7B N1B N1B N3B N3B HN31 N3B HN32
BOND N1B C8B C8B N2B
BOND N2B C1 N2B HN2B
BOND C7B O7B C8B O8B
BOND C1 C7C C7C N1C
BOND N1C C9C C9C H91 C9C H92 C9C H93
BOND N1C C8C C8C N2C
BOND N2C C1 N2C HN2C
BOND C7C O7C C8C O8C
IMPR C7 C1 N1 O7 N1 C7 C8 HN1 C8 N1 N2 O8
IMPR N2 C1 C8 HN2
IMPR C7B C1 N1B O7B N1B C7B C8B N3B C8B N1B N2B O8B
IMPR N2B C1 C8B HN2B N3B N1B HN31 HN32 N3B N1B HN32 HN31
IMPR C7C C1 N1C O7C N1C C7C C8C C9C C8C N1C N2C O8C
IMPR N2C C1 C8C HN2C
DONO BLNK HO2
DONO BLNK HO3
DONO BLNK HO4
DONO BLNK HO6
DONO BLNK HN1
DONO BLNK HN2
DONO BLNK HN2B
DONO BLNK HN2C
DONO BLNK HN31
DONO BLNK HN32
ACCE O2
ACCE O3
ACCE O4
ACCE O5
ACCE O6
ACCE O7
ACCE O8
ACCE O7B
ACCE O8B
ACCE O7C
ACCE O8C
PATC FIRS NONE LAST NONE
!
! patch residue to delete the angles
! N2-C1-N2B
! C7-C1-C7B
! N2-C1-N2C
! C7-C1-C7C
! N2B-C1-N2C
! C7B-C1-C7C
!
PRES DEL2 0.00
DELE ANGLE N2 C1 N2B
DELE ANGLE N2 C1 C7B
DELE ANGLE C7 C1 C7B
DELE ANGLE C7 C1 N2B
DELE ANGLE N2 C1 N2C
DELE ANGLE N2 C1 C7C
DELE ANGLE C7 C1 C7C
DELE ANGLE C7 C1 N2C
DELE ANGLE N2B C1 N2C
DELE ANGLE N2B C1 C7C
DELE ANGLE C7B C1 C7C
DELE ANGLE C7B C1 N2C
ATOM, the second column
list the names of the atoms, the third are the types of the atoms,
and the fourth are the charges. Atoms that are grouped together with
a GROU tag may receive some sort of treatment for electrostatic
interactions. Lines that start with BOND define the bonds.
Lines that start with IMPR define the improper dihedral angles.
Lines that start with DONO and ACCE
list the hydrogen bond donors and acceptors.
A B 305.000 1.3750 ! A-B BOND, by Blah Blah Blah
The first number is the bond stretching constant, the second number
is the bondlength. In the Charmm Hamiltonian, a bond is represented
by a harmonical potential which restraints the distance between the
two bond participants to be around the bondlength with a large force
constant. The characters after the exclamation are regarded as the
explanation field. If the user wants to add an angle, add a line like
this in the ANGLES category
A B C 48.00 123.50 ! A-B-C Angle, by Blah Blah Blah
The first number is the angle bending force constant, the second number is
the angle which it will be restrained to.
If the user wants to add a dihedral angle, add a line like
this in the DIHEDRALS category
A B C D 0.2000 1 180.00 ! A-B-C-D Dihedral, by Blah Blah Blah
The first number is the barrier height for the torsional potential,
the second is the multiplicity, which gives the number of minima in
the function as the bond is rotated through 360 degree, the third
is the phase factor that determines where the torsion angle passes
through its minimum value. Improper dihedrals are defined in a similiar
way except that they should be put in the IMPROPER category
and have zero multiplicity.
Write a Charmm script for MD simulation
The following is the template script for performing molecular
dynamics simulation for azurin in either vacuum or solution.
This script tries
to automate the job with a single main file plus a few plug-in inputs
(the so-called 'streams' in Charmm), starting from experimental
protein structure(azuh.pdb---the crystallographic structure
for azurin augmented with hydrogen atoms built using HBUILD,
and wath.pdb---the file containing crystallographic waters).
Azurin is a small protein, so no cutoff procedure is needed to downsize
the system.
The switches are summarized as follows:
Restart: 1/0---restart from history/start from scratch;
Solvent: 1/0---with/without solvent;
CryWater: 1/0---intake/ignore crystallographic waters;
RefillWater: 1/0---add/do not add more waters;
RefillRestart: 1/0---restart after adding more waters/do not restart;
FixProtein: 1/0---fix protein/do not fix protein;
run: 1/0---run/do not run a MD simulation;
TimeSeries: 1/0---create/do not create time series;
WritePDB: 1/0---write/do not write PDB series;
Minimize: 1/0---minimize/do not minimize the structure;
Switch1: composite switch;
Switch2: composite switch;
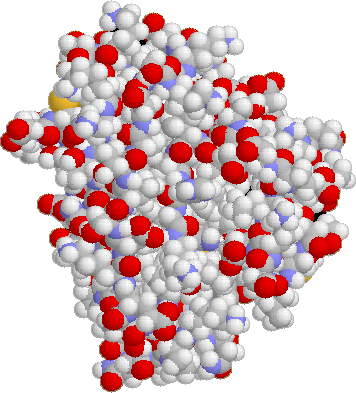
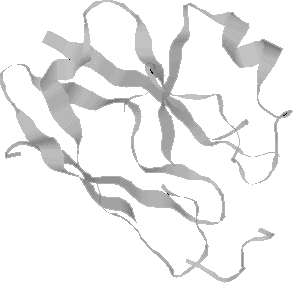
The azurin molecule: rendered in space-filled and ribbon display modes.
Images created by Rasmol and Photoshop.
Solvent
and CryWater. When the user wishes to add more waters to the
system, he can simply turn on the RefillWater. After this
step, he has to turn it off and turn on the RefillRestart
switch in order to continue the simulation. If he mistakenly turns on
the Restart, the script will refuse to work as Charmm will
detect that the total number of atoms has been changed and exit.
* TITLE Molecular Dynamics for Azurin
*
set Restart 1
set RefillWater 0
set RefillRestart 0
set run 1
set Solvent 1
set FixProtein 0
set CryWater 1
set NumberofSteps 50
set Steplength 0.001
set freqsave 1
set trajskip 1
set Temperature 300.0
set Timeseries 0
set WritePDB 1
set NumberofPDB 50
set Minimize 0
set minstep 100
set tolfun 0.0001
set Radius 28
set OverlayDiam 2.8
BOMLEV -1
set Switch1 0
set Switch2 0
set Start 0
let Start = 1 - @Restart
let Switch1 = @Refillwater * @Restart
let Switch2 = @Start * @Solvent
set fact 0.0
set IndexofOverlay 0
set xphi 1.0
set yphi 0.0
set zphi 0.0
set phi 0.0
set xdir 0.0
set ydir 0.0
set zdir 0.0
set MainDir /home/xie/charmm
set WaterDir /home/xie/charmm/bpot
set PDBDir /home/xie/strk/pdb
set rstfi @MainDir/scratch/azurin.rst
set dcdfi @MainDir/scratch/azurin.dcd
set velfi @MainDir/scratch/azurin.vel
set dcdor @MainDir/scratch/azurinorie.dcd
open read card unit 10 name @MainDir/toppar/top_all22_prot_na.inp
read rtf card unit 10
open read card unit 10 name @MainDir/toppar/par_all22_prot_na2.inp
read para card unit 10
label BEGIN
if RefillWater eq 1 goto BUILD1
if RefillRestart eq 1 goto BUILD4
if Restart eq 1 goto BUILD1
open read card unit 10 name @MainDir/azurin/azuh.pdb
read sequence pdb unit 10
generate AZUA setup warn first NTER last CTER
PATCH DISU AZUA 3 AZUA 26
patch HSQP AZUA 46 AZUA 117
patch HSQC AZUA 46 AZUA 112
open read card unit 10 name @MainDir/azurin/azuh.pdb
read coor pdb unit 10 sele segid AZUA end
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/wath.pdb
if CryWater eq 1 read sequence pdb unit 10
if CryWater eq 1 generate WATE noangle nodihedral setup warn
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/wath.pdb
if CryWater eq 1 read coor pdb sele segid WATE end unit 10
if Restart eq 0 goto BUILD2
label BUILD1
open read card unit 10 name @MainDir/azurin/azurin.pdb
read sequence pdb unit 10
generate AZUA setup warn first NTER last CTER
PATCH DISU AZUA 3 AZUA 26
patch HSQP AZUA 46 AZUA 117
patch HSQC AZUA 46 AZUA 112
open read card unit 10 name @MainDir/azurin/azurin.pdb
read coor pdb unit 10 sele segid AZUA end
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/crywat.pdb
if CryWater eq 1 read sequence pdb unit 10
if CryWater eq 1 generate WATE noangle nodihedral setup warn
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/crywat.pdb
if CryWater eq 1 read coor pdb sele segid WATE end unit 10
if Solvent eq 1 open read card unit 30 name water.pdb
if Solvent eq 1 read sequence pdb unit 30
if Solvent eq 1 generate SOLV noangle nodihe setup warn
if Solvent eq 1 read coor pdb unit 30 sele segid SOLV end
label BUILD2
! Overlay water, rotate more, and overlay again
if Switch1 eq 0 goto BUILD3
stream azurin.rfl.str
incr IndexofOverlay by 1
incr phi by 90
if IndexofOverlay lt 2 goto BUILD2
STOP
label BUILD3
goto BUILD5
label BUILD4
open read card unit 10 name @MainDir/azurin/azurin.pdb
read sequence pdb unit 10
generate AZUA setup warn first NTER last CTER
PATCH DISU AZUA 3 AZUA 26
patch HSQP AZUA 46 AZUA 117
patch HSQC AZUA 46 AZUA 112
open read card unit 10 name @MainDir/azurin/azurin.pdb
read coor pdb unit 10 sele segid AZUA end
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/crywat.pdb
if CryWater eq 1 read sequence pdb unit 10
if CryWater eq 1 generate WATE noangle nodihedral setup warn
if CryWater eq 1 open read card unit 10 name @MainDir/azurin/crywat.pdb
if CryWater eq 1 read coor pdb sele segid WATE end unit 10
open read card unit 30 name water.pdb
read sequence pdb unit 30
generate SOLV noangle nodihe setup warn
read coor pdb unit 30 sele segid SOLV end
let Restart = 0
let Start = 1
label BUILD5
! modify the charges of the copper ligands
.
.(ignored here, check azurin.main for details)
.
! =============================================
scalar wmain = charge sele all end
coor statistics sele all end
set TotalCharge ?WAVE
Multiply TotalCharge by ?NATO
! define and constrain the improper dihedrals for the copper complex
ic edit
dihe AZUA 46 ND1 AZUA 112 SG AZUA 46 CU AZUA 117 ND1 180.0
dihe AZUA 112 SG AZUA 117 ND1 AZUA 46 CU AZUA 46 ND1 180.0
dihe AZUA 117 ND1 AZUA 46 ND1 AZUA 46 CU AZUA 112 SG 180.0
end
ic fill
cons dihe AZUA 46 ND1 AZUA 112 SG AZUA 46 CU AZUA 117 ND1 force 10.0 min 180.0
cons dihe AZUA 112 SG AZUA 117 ND1 AZUA 46 CU AZUA 46 ND1 force 10.0 min 180.0
cons dihe AZUA 117 ND1 AZUA 46 ND1 AZUA 46 CU AZUA 112 SG force 10.0 min 180.0
if RefillRestart eq 1 goto MAIN001
if Switch2 eq 1 stream azurin.ini.str
label MAIN001
if Solvent eq 1 stream azurin.set.str
if run eq 1 stream azurin.dyn.str
open write card unit 10 name @MainDir/azurin/total.pdb
write coor pdb unit 10 sele all end
if Solvent eq 1 open write card unit 10 name @MainDir/azurin/water.pdb
if Solvent eq 1 write coor pdb unit 10 sele segid SOLV end
open write card unit 10 name @MainDir/azurin/azurin.pdb
write coor pdb unit 10 sele segid AZUA end
if CryWater eq 1 open write card unit 10 name @MainDir/azurin/crywat.pdb
if CryWater eq 1 write coor pdb unit 10 sele segid WATE end
if WritePDB eq 1 stream azurin.pdb.str
if Timeseries eq 1 stream azurin.tim.str
stop
Postprocess a MD simulation
Prune the structure and create the PDB series
The plug-in file (
azurin.pdb.str) truncates part of the beta-sheet from
the whole azurin molecule. When installing ACE and
CT3 terminals
to the two ends of both strands, care is taken such that the terminal atoms
sit on the positions of the corresponding atoms of the excluded residues that
are right next to the end residues. This can be obviously seen from the
following script.
The reason that we add ACE and CT3 terminals
is that they satisfy the close-shell requirement for the electronic
structure calculation (CNDO level).
* extract the pathway from the whole protein and patch ACE and CT3 to the
* two ends of the pathway
OPEN READ FILE UNIT 61 NAME @dcdfi
TRAJ IREAD 61 NREAD 1 SKIP @trajskip BEGIN 000 STOP 5000000
SET INDEX 1
LABEL CONT
TRAJ READ
OPEN WRITE CARD UNIT 60 NAME pathway1.pdb
WRITE COOR PDB UNIT 60 sele (segid AZUA .and. resid 121:123) end
open write card unit 60 name pathway2.pdb
write coor pdb unit 60 sele (segid AZUA .and. resid 110:112) end
coor statistics sele atom AZUA 120 C end
set xC120 ?xave
set yC120 ?yave
set zC120 ?zave
coor statistics sele atom AZUA 120 CA end
set xCA120 ?xave
set yCA120 ?yave
set zCA120 ?zave
coor statistics sele atom AZUA 120 O end
set xO120 ?xave
set yO120 ?yave
set zO120 ?zave
coor statistics sele atom AZUA 124 N end
set xN124 ?xave
set yN124 ?yave
set zN124 ?zave
coor statistics sele atom AZUA 124 CA end
set xCA124 ?xave
set yCA124 ?yave
set zCA124 ?zave
coor statistics sele atom AZUA 124 HN end
set xHN124 ?xave
set yHN124 ?yave
set zHN124 ?zave
coor statistics sele atom AZUA 109 C end
set xC109 ?xave
set yC109 ?yave
set zC109 ?zave
coor statistics sele atom AZUA 109 CA end
set xCA109 ?xave
set yCA109 ?yave
set zCA109 ?zave
coor statistics sele atom AZUA 109 O end
set xO109 ?xave
set yO109 ?yave
set zO109 ?zave
coor statistics sele atom AZUA 113 N end
set xN113 ?xave
set yN113 ?yave
set zN113 ?zave
coor statistics sele atom AZUA 113 CA end
set xCA113 ?xave
set yCA113 ?yave
set zCA113 ?zave
coor statistics sele atom AZUA 113 HN end
set xHN113 ?xave
set yHN113 ?yave
set zHN113 ?zave
rename segid TEMP sele segid AZUA end
open read card unit 60 name pathway1.pdb
read sequence pdb unit 60
generate AZUA setup warn first ACE last CT3
read coor pdb unit 60
scalar x set @xC120 sele (segid AZUA .and. resname MET .and. type CY) end
scalar y set @yC120 sele (segid AZUA .and. resname MET .and. type CY) end
scalar z set @zC120 sele (segid AZUA .and. resname MET .and. type CY) end
scalar x set @xCA120 sele (segid AZUA .and. resname MET .and. type CAY) end
scalar y set @yCA120 sele (segid AZUA .and. resname MET .and. type CAY) end
scalar z set @zCA120 sele (segid AZUA .and. resname MET .and. type CAY) end
scalar x set @xO120 sele (segid AZUA .and. resname MET .and. type OY) end
scalar y set @yO120 sele (segid AZUA .and. resname MET .and. type OY) end
scalar z set @zO120 sele (segid AZUA .and. resname MET .and. type OY) end
scalar x set @xN124 sele (segid AZUA .and. resname GLY .and. type NT) end
scalar y set @yN124 sele (segid AZUA .and. resname GLY .and. type NT) end
scalar z set @zN124 sele (segid AZUA .and. resname GLY .and. type NT) end
scalar x set @xCA124 sele (segid AZUA .and. resname GLY .and. type CAT) end
scalar y set @yCA124 sele (segid AZUA .and. resname GLY .and. type CAT) end
scalar z set @zCA124 sele (segid AZUA .and. resname GLY .and. type CAT) end
scalar x set @xHN124 sele (segid AZUA .and. resname GLY .and. type HNT) end
scalar y set @yHN124 sele (segid AZUA .and. resname GLY .and. type HNT) end
scalar z set @zHN124 sele (segid AZUA .and. resname GLY .and. type HNT) end
hbuild
rename segid PAR1 sele segid AZUA end
! hack here: since we cannot do open-shell calculations, charges are
! removed, and sulfur atoms replaced by oxygen atoms.
delete atom sele (segid PAR1 .and. resname LYS .and. type HZ3) end
rename atom OD sele (segid PAR1 .and. resname MET .and. type SD ) end
label cont200
open read card unit 60 name pathway2.pdb
read sequence pdb unit 60
generate AZUA setup warn first ACE last CT3
read coor pdb unit 60
scalar x set @xC109 sele (segid AZUA .and. resname PHE .and. type CY) end
scalar y set @yC109 sele (segid AZUA .and. resname PHE .and. type CY) end
scalar z set @zC109 sele (segid AZUA .and. resname PHE .and. type CY) end
scalar x set @xCA109 sele (segid AZUA .and. resname PHE .and. type CAY) end
scalar y set @yCA109 sele (segid AZUA .and. resname PHE .and. type CAY) end
scalar z set @zCA109 sele (segid AZUA .and. resname PHE .and. type CAY) end
scalar x set @xO109 sele (segid AZUA .and. resname PHE .and. type OY) end
scalar y set @yO109 sele (segid AZUA .and. resname PHE .and. type OY) end
scalar z set @zO109 sele (segid AZUA .and. resname PHE .and. type OY) end
scalar x set @xN113 sele (segid AZUA .and. resname CYS .and. type NT) end
scalar y set @yN113 sele (segid AZUA .and. resname CYS .and. type NT) end
scalar z set @zN113 sele (segid AZUA .and. resname CYS .and. type NT) end
scalar x set @xCA113 sele (segid AZUA .and. resname CYS .and. type CAT) end
scalar y set @yCA113 sele (segid AZUA .and. resname CYS .and. type CAT) end
scalar z set @zCA113 sele (segid AZUA .and. resname CYS .and. type CAT) end
scalar x set @xHN113 sele (segid AZUA .and. resname CYS .and. type HNT) end
scalar y set @yHN113 sele (segid AZUA .and. resname CYS .and. type HNT) end
scalar z set @zHN113 sele (segid AZUA .and. resname CYS .and. type HNT) end
hbuild
rename segid PAR2 sele segid AZUA end
! hack here: since we cannot do open-shell calculations, charges are
! removed, and sulfur atoms replaced by oxygen atoms.
rename atom OG sele (segid PAR2 .and. resname CYS .and. type SG ) end
rename atom OD sele (segid PAR2 .and. resname MET .and. type SD ) end
OPEN WRITE CARD UNIT 60 NAME @PDBDir/@INDEX.pdb
WRITE COOR PDB UNIT 60 sele segid PAR1 .or. segid PAR2 end
delete atom sele segid PAR1 .or. segid PAR2 end
rename segid AZUA sele segid TEMP end
INCR INDEX BY 1
IF INDEX GT @NumberofPDB GOTO EXIT
GOTO CONT
LABEL EXIT
 |
A C-shell script loop-runs the above Charmm script, and deposits the PDB files into a series of segments.
#!/bin/csh -f # script file for md and saving pdb files #initialize pwd date @ number_of_run = 200 @ length_of_run = 50 @ i0 = 0 @ i = $i0 set dir_of_md = /home/xie/charmm set a = `pwd` if ( $a != $dir_of_md ) then echo We are not in the right starting directory! exit endif cd ./azurin/ label_one: @ i ++ runch < azurin.main cd cd ./strk/pdb if ( -e 1.pdb ) then tar -cvf azur.$i.tar *.pdb gzip azur.$i.tar mv azur.$i.tar.gz /home/xie/strk/ rm -f *.pdb endif cd ../../charmm/azurin/ if ( $i - $i0 < $number_of_run ) goto label_one date pwd exitSome DCD trajectories for the beta-strands and beta-sheet are stored in a directory on thales.